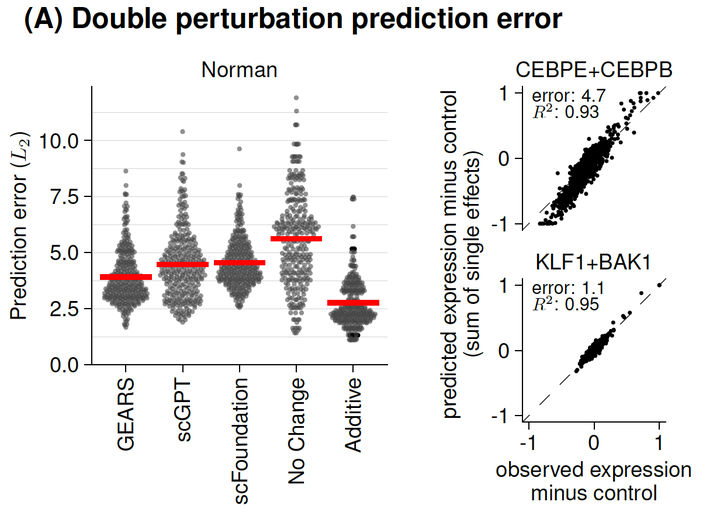
Deep learning-based predictions of gene perturbation effects do not yet outperform simple linear methods
 Prediction error
Prediction error
Abstract
Advanced deep-learning methods, such as transformer-based foundation models, promise to learn representations of biology that can be employed to predict in silico the outcome of unseen experiments, such as the effect of genetic perturbations on the transcriptomes of human cells. To see whether current models already reach this goal, we benchmarked two state-of-the-art foundation models and one popular graph-based deep learning framework against deliberately simplistic linear models in two important use cases: For combinatorial perturbations of two genes for which only data for the individual single perturbations have been seen, we find that a simple additive model outperformed the deep learning-based approaches. Also, for perturbations of genes that have not yet been seen, but which may be “interpolated” from biological similarity or network context, a simple linear model performed as good as the deep learning-based approaches. While the promise of deep neural networks for the representation of biological systems and prediction of experimental outcomes is plausible, our work highlights the need for critical benchmarking to direct research efforts that aim to bring transfer learning to biology.