Comparison of transformations for single-cell RNA-seq data
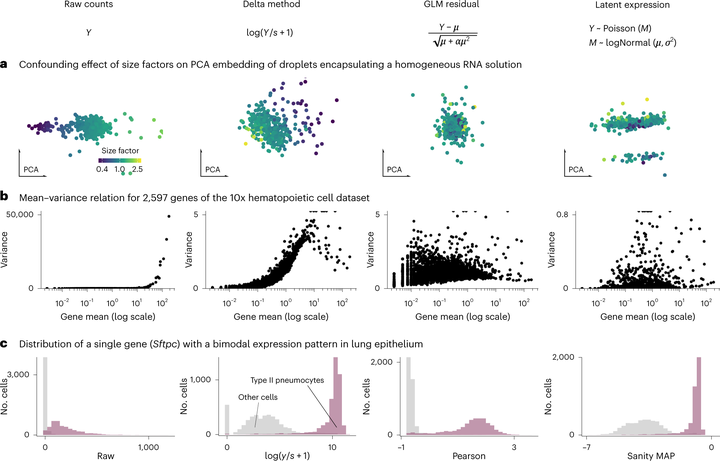 Conceptual differences
Conceptual differences
Abstract
The count table, a numeric matrix of genes × cells, is the basic input data structure in the analysis of single-cell RNA-sequencing data. A common preprocessing step is to adjust the counts for variable sampling efficiency and to transform them so that the variance is similar across the dynamic range. These steps are intended to make subsequent application of generic statistical methods more palatable. Here, we describe four transformation approaches based on the delta method, model residuals, inferred latent expression state and factor analysis. We compare their strengths and weaknesses and find that the latter three have appealing theoretical properties; however, in benchmarks using simulated and real-world data, it turns out that a rather simple approach, namely, the logarithm with a pseudo-count followed by principal-component analysis, performs as well or better than the more sophisticated alternatives. This result highlights limitations of current theoretical analysis as assessed by bottom-line performance benchmarks.